

Différence entre amyloïde et prion
- 2806
- 180
- Justine Dumas
Les amyloïdes et les prions sont tous deux synonymes de troubles neurodégénératifs, troubles qui provoquent une dégénérescence dans le cerveau. Les amyloïdes ne sont pas spécifiques au cerveau, car ils peuvent provoquer des troubles dans les organes du corps. En termes de neurologie, les troubles neurodégénératifs humains pour lesquels les amyloïdes et les prions sont directement ou indirectement responsables sont ceux tels que la maladie d'Alzheimer, la maladie de Parkinson et la maladie de Creutzfeldt-Jakob. La compréhension du fonctionnement au sein des amyloïdes constitue un aspect important de la compréhension des prions et de la neurodégénérescence.
Différence entre amyloïde et prion
Définition et physiopathologie
Les amyloïdes sont des agrégats (une formation en plusieurs parties compilées ensemble) d'une structure en forme de tige qui contient des répétitions d'un certain type de protéine. Lorsque des plasmocytes anormaux provenant de la moelle osseuse forment des protéines de chaîne légère anormales, elles pénètrent dans la circulation sanguine et forment des dépôts amyloïdes. Ces dépôts peuvent provoquer une accumulation dans n'importe quel organe vital du corps (comme le cerveau, le cœur ou les intestins), résultant en diverses conditions médicales graves.
Les prions sont une forme ou un pliage anormal des protéines spécifiques des amyloïdes déposées dans le cerveau, ce qui les rend infectieuses et capables de renouveler indéfiniment. En d'autres termes, les prions sont définies comme une sous-classe d'amyloïdes où l'agrégation des protéines est venue infectieuse et a changé l'état d'auto-production. Les prions ont la capacité de transmettre leur forme de protéine mal repliée en les mêmes protéines se produisant dans un état normal.
Les prions sont responsables des troubles neurodégénératifs qui peuvent survenir chez l'homme et les animaux. Cela est dû au fait que la protéine déformée est plus difficile à décomposer par les enzymes et finir par s'accumuler dans les neurones à la place, entraînant une destruction. Lorsque cette destruction de neurones progresse, elle fait progressivement les tissus du cerveau à avoir un motif en forme d'éponge rempli de trous.
Causes potentielles
Les amyloïdes se produisent lorsque la moelle osseuse produit des plasmocytes sanguins anormaux. Ces plasmocytes anormaux forment alors des types anormaux de protéines de chaîne légère. Lorsque les protéines anormales de chaîne légère pénètrent dans le système de circulation (circulation sanguine), ils se déposent dans des organes vitaux dans tout le corps. Cela peut être dû à:
- Composantes génétiques ou héréditaires (qui affectent les yeux, le cœur, les reins et le cerveau)
- Maladies chroniques et types de cancers
- Mutations cellulaires
- Infections rares
Les prions sont les protéines déformées provoquant des amyloïdes déposés dans le cerveau. Génétiquement, le gène PRNP est chargé de diriger le corps vers la protéine de prion produit. Lorsque ce gène mute, il peut provoquer une production anormale de protéines, entraînant des maladies à prion spécifiques du cerveau. La maladie à prion peut également se produire sporadiquement, entraînant d'autres maladies spécifiques du cerveau.
Symptômes
Les symptômes des dépôts de protéines amyloïdes anormaux varient et dépendent du tissu ou de l'organe affecté par les dépôts.
La maladie à prion, causée par des protéines prions difformes, entraînant des amyloïdes dans le cerveau, présente souvent les symptômes suivants:
- Difficultés de mémoire et changements de jugement
- Changements de personnalité
- Confusion, difficulté de parole et / ou désorientation
- Changements de coordination et de spasmes musculaires involontaires
- Diminution de la qualité de vision ou de la cécité
Diagnostic
Le diagnostic de l'occurrence d'amyloïdes dans les organes affectés peut-être spécifiquement peut être effectué par le biais de tests de biomarqueurs. Il s'agit notamment de mesurer les changements de taille de l'organe et de ses fonctions, de mesurer les niveaux de protéines spécifiques dans le sang, dans le liquide céphalorachidien et sur les scans (IRM, CT, PET).
Le diagnostic des protéines prions dans le cerveau provoquant une maladie à prions peut être effectuée par des analyses du cerveau à mesurer la fonction et la taille, les tests de liquide céphalo-rachidien pour les marqueurs spécifiques de la maladie de prion et les marqueurs de neurodégénérescence, et l'EEG à enregistrer les changements d'activité électrique dans le cerveau.
Traitement
Le traitement des amyloïdes dépend de l'endroit où ils se produisent. La seule façon d'abolir complètement les amyloïdes de la production est de subir une chimiothérapie intensive pour détruire les cellules sanguines anormales dans la moelle osseuse responsable de la production amyloïde. Il n'y a aucun autre moyen de traiter les amyloïdes et les troubles causés par leur occurrence sont traités sur une base spécifique.
Les prions provoquant une maladie à prions peuvent être traités avec des médicaments, une vie assistée associée à la neurodégénérescence, à l'hydratation de maintien et à l'apport en nutriments. Cela ne guérit pas la production de prions ou de neurodégénérescence, mais fournit un moyen d'améliorer la qualité de vie et de traiter les symptômes associés.
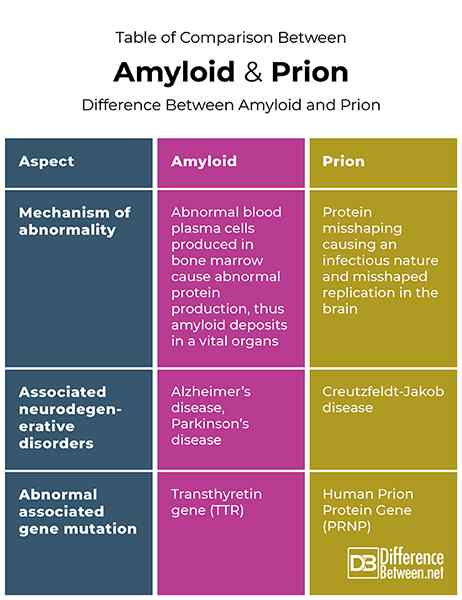
Tableau de comparaison entre amyloïde et prion
Résumé de l'amyloïde vs prion
Les amyloïdes et les prions sont tous deux associés à la démission des protéines et à certains troubles neurodégénératifs. Dans le cas des amyloïdes, les protéines difficultées peuvent provoquer des troubles dans n'importe quel organe où ils sont déposés. Les prions, cependant, sont les protéines déformées qui peuvent provoquer des amyloïdes dans le cerveau et ont des troubles associés limités au cerveau. En termes de troubles neurodégénératifs, les amyloïdes sont responsables de la maladie d'Alzheimer et de la maladie de Parkinson, où les prions sont responsables de la maladie de Creutzfeldt-Jakob.
FAQ
Les prions sont-ils un type d'amyloïde?
Les amyloïdes sont des agrégats avec une structure fibrillaire qui est constituée par les répétitions d'une protéine spécifique. Les prions sont définis comme un sous-classe d'amyloïdes. Avec les prions, l'agrégation des protéines devient auto-perpétuante puis devient infectieuse - c'est la cause de nombreuses maladies neurodégénératives mortelles telles que la maladie de Creutzfeldt-Jacob.
Comment les plaques amyloïdes sont-elles liées aux prions?
Des plaques amyloïdes ont été trouvées dans le cerveau des personnes atteintes de la maladie de Creutzfeldt-Jacob. La maladie de Creutzfeldt-Jacob est une maladie causée par les prions. La ferbloriation est une autre maladie à prion, cette fois trouvée chez les animaux, avec des plaques amyloïdes qui sont composées de protéines prions
Quelle est la différence entre un prion et une protéine?
La principale différence entre une protéine et un prion est dans la structure - les prions sont mal repliés les obligeant à changer la forme d'autres protéines similaires, devenant ainsi infectieuse.
Est amyloïde bêta un prion?
En raison de sa capacité à s'auto-copier ainsi qu'à l'existence de plusieurs «souches» distinctes, la bêta amyloïde partage de nombreuses propriétés indiscernables avec des prions. Ces propriétés sont ce qui permet probablement de devenir un prion pendant les maladies.
- « Différence entre le syndrome du côlon irritable et l'intolérance au lactose
- Différence entre la force électromotive et la force magnétomotive »